




Gaussian - 量子化学综合软件包
美国Gaussian公司与2019年7月24日发布了Gaussian 16 Revision C.01
Gaussian软件是目前计算化学领域内应用范围广的综合性量子化学计算程序包,广泛的被各地的化学家、化学工程师、生物化学家、物理学家和学科的科学家们使用。Gaussian软件基于量子力学而开发,它致力于把量子力学理论应用于实际问题,它可以通过一些基本命令验证和预测目标体系几乎质。其可执行程序可在不同型号的大型计算机,计算机,工作站和个人计算机上运行,并相应有不同的版本。此外,可视化软件GaussView的发布及计算机的发展更是大大降低了理论计算的门槛,使得各领域研究者能够使用Gaussian研究和分析科学问题。
Gaussian能够过渡态能量和结构、键和反应能量、分子轨道、原子电荷的电势、振动频率、红外和拉曼光谱、核磁性质、级化率和化率、热力学性质、反应路径,计算可以对体系的基态或激发态执行。可以预测周期体系的能量,结构和分子轨道。因此,Gaussian可以作为功能的工具,用于研究化学领域的课题,例如取代基的影响,化学反应机理,势能曲面和激发能等等。
Gaussian 16从量子力学的基本定律出发,在不同的化学环境中预测分子结构、能量、振动频率、分子性质与反应。Gaussian 16的模型既可以应用于稳定的体系与化合物,也适用于实验中很难或不可能观察到的体系或化合物(例如,生存周期很短的中间体和过渡态结构)。Gaussian 16提供了当今所能得到的建模功能。它新功能和增强功能,其的扩大了可以研究的问题和体系的尺度。通过Gaussian 16,你可以在计算硬件条件下研究以往时候都要大的多的体系以及更复杂的问题。
Gaussian 16 C.01版本的Linda版需要将Linda到TCP-Linda 9.2。
Gaussian 16 带来了新方法,属性预测和性能增强。
激发态建模
时变(TD)Hartree-Fock和DFT方法的解析频率计算,与MM区域环境耦合的ONIOM电子嵌入,近似方法。
使用高精度EOM-CCSD方法(解读梯度)进行几何
用于计算IR,拉曼,VCD和ROA光谱的分析。溶液中的计算充分考虑了激发和溶剂场之间的相互作用。
振动光谱预测
手性光谱学:电子圆二色性(ECD)和圆偏振发光
共振拉曼光谱建模
电子能量转移(EET)的计算
Ciofini的激发电荷转移诊断
Caricato的EOM-CCSD溶剂化相互作用模型
新方法
自G09发布以来,DFT功能已添加到Gaussian函数中,APFD,Truhlar组的功能
双杂交方法:DSDPBEP86,PBE0DH,PBEQIDH
使用Grimme(GD2,GD3,GD3BJ)等方案对功能进行经验分散
PM7半经验方法,既有原始公式,也有对连续势能面的修改
性能增强
大量处理器的并行计算
当跨10到20多个内核运行时,Gaussian 16能明显优于以前的版本。丙氨酸25以上的APFD / 6-31G(d)Opt=CalcFreq计算在Gaussian 16中比在Gaussian 09中快1.8倍(参见以下图表)。计算的CalcFC和Freq部分在Gaussian 16中占执行时间47%,而在Gaussian 09中为61%。
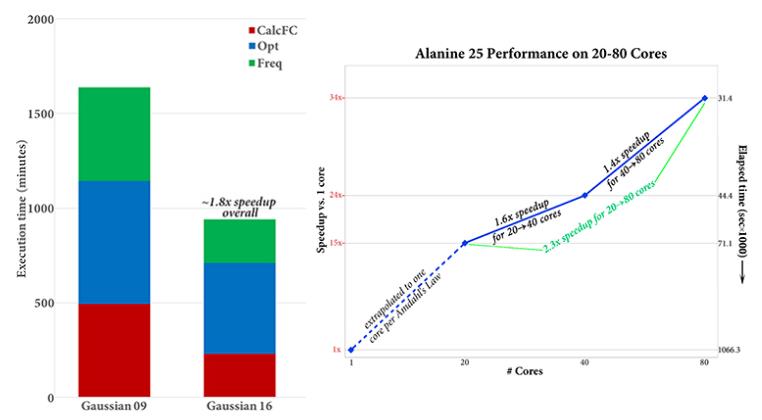
Gaussian 16 能可扩展到80核心或更多(取决于计算和分子)。右上方的图表给出了在Alanine 25上进行APFD / 6-31G(d)Freq计算时20、40和80核的加速,这项工作显示出随着处理器数量的增加,并行速度得到了很好的提高。从20核迁移到80核时的加速比约为2.3,而在单个处理器上估计80核的加速比为34.
在Linux系统上NVIDIA K40和K80 GPU,用于HF和DFT计算
加快了几种计算类型的关键部件的速度
CASSCF的改进和(不少于)16个轨道的活动空间
算法上的改进
Gaussian 16还为程序的不同部分提供了多种性能的改进。其中:
-
一种的存储算方法,可避免CCSD轶代期间的I / O
-
GEDIIS功能的增强提供了的,并且对相应算法进行了更复杂的自动检测/选择
-
CASSCF的改进允许使用的空间
-
EPT(OVGF)复合模型的低水平部分用于预测光电子光谱能要快的多
易于使用的功能
-
Gaussian 16 Opt(Recalc=n)在几何的第n个步骤中,使用的分析值自动更新力常数。这对于在过程中发生较大的分子系统很有帮助。
-
Link 0命令的扩展集:受的指令集提供了对计算资源使用情况的直接管理。Link 0 命令在Default.route文件伪指令中都有对应的命令,因此可以在系统范围内指定和管理资源。他们都有新颖的命令行选项,因此脚本可以使用相同的功能。
-
Gaussian 与程序接口的工具,既可以使用Fortran和C等编译语言,也可以使用Python和Perl等解释语言。这些工具是根据Mozilla公共的变体提供的。他们是为以下情况而设计的:
-
自动运行多个相关Gaussian作业的脚本
-
Gaussian结果的后处理
-
程序中的计算可以使用由Gaussian生成的中间数据,方法是运行使用前计算的Gaussian结果的单个程序,或者将外部程序的结果合并到Gaussian计算中
-
通过使用由Gaussian计算的数量来建立新模型/方法的原型,从而需要实现新模型的奇特方面
-
通用内部表(GIC):可以定义任意冗余的内部坐标。GIC可以潜在地用于在分子系统过程中冻结葛洪结构参数,指定要执行扫描的参数以及基于结构参数或他们之间的复杂管理定义几何的约束
使用新的画笔选择工具,您可以在橡皮工具无法到达的秘籍区域中选择一组原子,这些原子对于单个单击而言太大。您还可以使用增强的原子选择工具按键,距离,PDB残基或定义的原子组件选择原子。
可以在步骤中对分子组中的原子或一组文件执行操作。其中设置Gaussian计算,保存输入文件,将作业提交到SC Job Manager,并将分子图像捕获为图形文件。
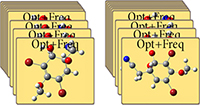
上面的对话框显示了多文件保存功能。我们使用它为分子组中结构的输入文件分配的文件名,然后将它们保存在操作中。
Gaussian软件能够研究诸多的科学问题,例如:
-
化学反应过程,如稳态及过渡态结构确定、反应热、反应能垒、反应机理及反应动力学等;
-
各类型化合物稳态结构的确定,如中性分子、自由基、阴、阳离子等;
-
谱图的验证及预测,如IR, Raman, NMR, UV/Vis, VCD, ROA, ECD, ORD, XPS, EPR, Franck-Condon及超精细光谱等;
-
分子性质,如静电势、偶级矩、布居数、轨道、键级、电荷、级化率、电子亲和能、电离势、自旋密度、电子转移、手性等;
-
热力学分析,如熵变、焓变、吉布斯自由能变、键能分析及原子化能等;
-
分子间相互作用,如氢键及范德华作用;
-
激发态,如激发态结构确定、激发能、跃迁偶级矩、荧光光谱、磷光光谱、势能面交叉研究等。


- 2026-07-29
- 2026-07-28
- 2026-07-26
- 2026-07-24
- 2026-07-23
- 2026-07-21

- 2026-08-05
- 2026-07-30
- 2026-07-29
- 2026-07-29
- 2026-07-29
- 2026-07-29








