



ProMass - 生物分子解卷积和报告软件
ProMass是一种自动生物分子解卷积和报告软件包,用于处理Thermo Scientific的Xcalibur或Waters MassLynx数据系统上采集的ESI / LC / MS数据或单个ESI质谱图。ProMass被设计用于高通量应用。
当用电喷雾(ESI)离子化时,大的生物分子主要是多电荷的。当以正离子模式分析时,由于碱性氨基酸和氨基末端,肽或蛋白质将在多个位点质子化。另一方面,在负离子模式下分析的寡核苷酸由于酸性磷酸骨干而在多个位点去质子化。这种多次充电通常一系列电荷状态的ESI质谱。这些复杂的光谱“解卷积”或“转化”,以便确定生物分子的不带电荷的质量。 ProMass为执行整个LC / MS数据集的解卷积提供了自动化平台。
ProMass从一开始就设计用于高通量应用。实际上,运行ProMass的正常方式是通过Xcalibur或MassLynx示例列表。因此,您可以运行样品列表,让ProMass在处理它们,或者在您方便的时候批量重新处理列表。
ProMass使用称为ZNova的新型去卷积算法,可产生无伪像的解卷积质谱图。 ZNova可用于处理生物分子(蛋白质,寡核苷酸,肽等)的数据。ZNova可以适应低电荷状态谱和低信噪比的数据。 ProMass自动地以大多数网页浏览器的易用和交互式格式报告结果。摘要页面汇总了多次运行的结果,并将其显示在单个超链接页面上,以便您浏览结果。目标质量允许您自动从LC / MS数据中搜索多个目标质量数。目标质量结果颜色编码,使您可以磨合可能需要进一步关注的样品。
ProMass HR通过合并来自Positive Probability Ltd(PPL)的全套算法,用于高分辨率数据处理的功能。Promass HR方法可以利用PPL算法,该算法允许对同位素分辨的质谱进行单同位素质量测定或对同位素未分辨的质谱进行的分辨率。通过与Promass标准版本相同的机制,可以自动实现PPL方法。结果也通过相同的易于使用和直观的基于Wed的输出显示。
功能:
ProMass允许您以高度协作的基于Web的格式处理和传达您的结果。
-
生物分子质谱自动去卷积以确定分子量:适用于蛋白质,多肽,寡核苷酸等。
-
适用于Thermo Xcalibur或Waters MassLynx数据系统的ProMass版本。
-
使用ZNova算法的无伪影解卷积,手动干预去除伪像峰值。
-
用Xcalibur处理方法处理LC / MS峰。
-
处理Windows剪贴板上的光谱数据。
-
自动化的基于Web的报告,总结报告,色谱图(UV和MS),原始质谱图和解卷积质谱图。
-
自动计算目标寡核苷酸和多肽的预期质量。
-
颜色编码的结果显示,汇总报告和样品盘查看器可以查看结果。
-
自动搜索多个目标群体,并以颜色编码的基于Web的摘要报告结果。
-
基于Excel的总结报告选项
-
从寡核苷酸或蛋白质序列自动识别末端或内部片段
-
鉴定来自多个寡核苷酸或蛋白质序列的片段(例如双链体)
-
指定修饰的寡核苷酸
-
“模糊”靶标物质,如PTM或蛋白质药物缀合物
-
根据反卷积峰强度和色谱图面积计算目标质量纯度估算值
-
XML输出
-
ProMass当前可用于Thermo Xcalibur,Waters MassLynx和SCIEX数据系统。数据系统正在开发中。
ProMass版本分为标准版和HR版本。ProMass HR版本包含标准版的全部功能,另外,HR版本还具备以下额外功能:
-
带有ReSpectTM算法的ReView PPL数据处理包
-
Deisotoping用于同位素分辨质谱的单同位素质量测定,以获得的质量度(例如,Orbitrap数据<3 ppm)。
-
质谱峰形建模,用于提高同位素未分辨质谱的分辨率
-
用于同位素未解析质谱的PPL电荷反褶积算法
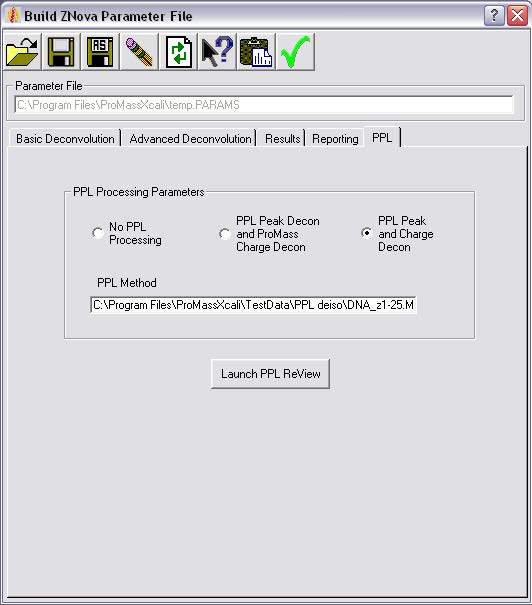

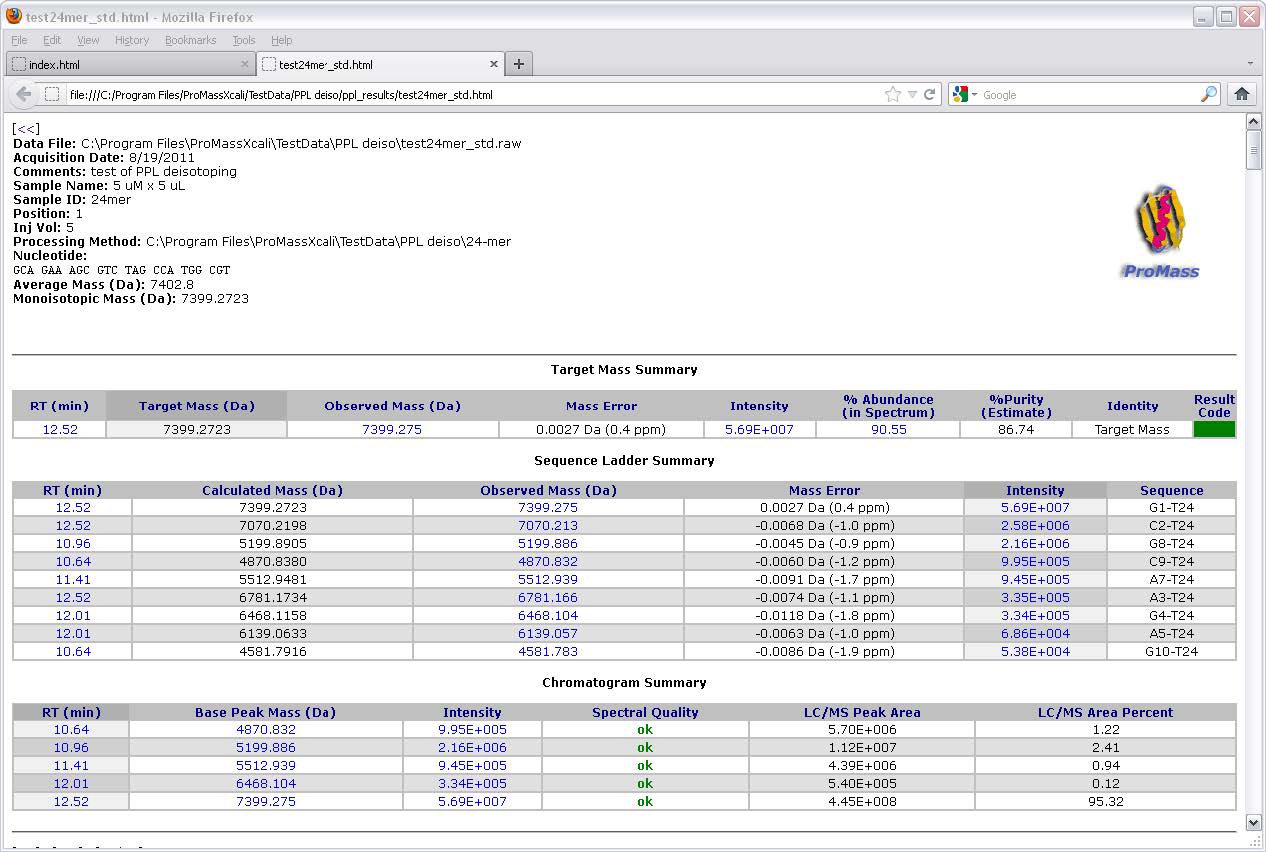
ProMass HR功能
-
ProMass HR包括用于高分辨率数据处理的功能
-
ProMass HR标准的ProMass反褶积算法和Positive Probability Ltd(PPL)数据处理包
-
PPL算法:去同位素(用于同位素分辨率质谱的单同位素质量测定)、峰值建模和形状反褶积,以及PPL电荷反褶积算法。
-
PPL峰形反褶积可与质子电荷反褶积相结合,以提高电荷反褶积前生物分子(如Mab)质谱的分辨率。这可以做到与低分辨率的数据。
-
ProMass HR可以引用ProMass参数文件,该文件嵌入了PPL方法来自动处理Xcalibur样本列表中的整个LCMS数据集。
-
ProMass HR使用与标准的ProMass反褶积包相同的易于使用的基于web的界面输出结果。
ProMass for Xcalibur系统要求
用户应该遵循安装计划使用的Xcalibur版本的系统要求。运行ProMass for Xcalibur的系统要求如下:
-
安装Thermo Xcalibur v.2.0.7或版本
-
应安装Thermo XDK(在Xcalibur CD中)以启用自动化功能
-
的PC与已安装的Thermo Xcalibur版本具有相同的要求
-
Microsoft Windows XP到Windows 10(受Xcalibur-Windows兼容性限制)
-
安装程序和帮助文件需要110 MB的可用磁盘空间
注意: ProMass不Web服务器软件。如果要在本地计算机上查看结果,则要Web服务器。但是,如果您想通过Internet或Intranet与他人共享数据,则需要Web服务器。
适用于Sciex系统要求的ProMass
您应该遵循Sciex OS的系统要求以实现Windows兼容性。下面列出了运行ProMass for Sciex的系统要求:
Windows 7或Windows 10
从Sciex获得ProMass Processor应用程序,以允许自动处理Sciex数据文件。
英文简介
Introduction:
Large biomolecules when ionized by electrospray (ESI) are predominantly multiply charged. When analyzed in the positive ion mode, peptides or proteins will be protonated at multiple sites as a result of the basic amino acids and amino terminus. On the other hand, oligonucleotides analyzed in the negative ion mode are de-protonated at multiple sites as a result of the acidic phosphate backbone. This multiple charging usually results in ESI mass spectra that contain a range of charge states. These complex spectra must be “deconvoluted” or “transformed” in order to order to determine the uncharged mass of the biomolecule. ProMass provides an automated platform for performing deconvolution of entire LC/MS data sets.
Overview:
ProMass is an automated biomolecule deconvolution and reporting software package that is used to process ESI/LC/MS data or single ESI mass spectra acquired on Thermo Scientific’s Xcalibur or Waters MassLynx data systems. ProMass was designed from the start for high-throughput applications. In fact, the normal way to run ProMass is through the Xcalibur or MassLynx sample list. Therefore, you can run a list of samples and have ProMass process them at the end or batch reprocess the list at your convenience.
ProMass uses a novel deconvolution algorithm known as ZNova that produces artifact-free deconvoluted mass spectra. ZNova can be used to process data from a wide variety of biomolecules including proteins, oligonucleotides, peptides, etc. Unlike many other deconvolution algorithms, ZNova can accommodate low charge state spectra and data of low signal-to-noise ratio. ProMass reports the results automatically in an easy-to-use and interactive format viewable from any web browser. A -level summary page summarizes the results from multiple runs and displays them on a single hyperlinked page allowing you to navigate your results quickly. Target mass features allow you to search for multiple target masses from LC/MS data automatically. The target mass features include results color coding, allowing you to quickly hone in on the samples that may require further attention.
ProMass HR includes additional features for high-resolution data processing by incorporating the full suite of algorithms from Positive Probability Ltd (PPL). ProMass HR methods can utilize the PPL algorithms that allow for monoisotopic mass determination of isotopically resolved mass spectra or enhanced resolution of isotopically unresolved mass spectra. The PPL methods may be automated thru the same powerful mechanism as the standard version of ProMass. Results are displayed through the same easy-to-use and intuitive web-based output as well.
Key Features of ProMass:
ProMass is more than just deconvolution software. ProMass is a tool that allows you to process and communicate your results in a highly collaborative web-based format.
-
Automated deconvolution of biomolecule mass spectra to determine molecular mass: applicable to proteins, peptides, oligonucleotides, etc.
-
ProMass versions for either Thermo Xcalibur or Waters MassLynx data systems
-
Artifact-free deconvolution using proprietary ZNova algorithm, no manual intervention required to remove artifact peaks
-
Flexible processing of LC/MS peaks directed by Xcalibur processing method
-
Process data from a spectrum on the Windows clipboard
-
Automated web-based reporting, including summary report, chromatograms (UV and MS), raw, and deconvoluted mass spectra
-
Automatically calculate expected masses of target oligonucleotides and polypeptides
-
Color-coded results display, top-level summary report, and sample plate viewer allow for rapid visualization of your results
-
Automatically search for multiple target masses and report results in color-coded web-based summary
-
Excel-based summary report option
-
Automatic identification of terminal or internal fragments from oligonucleotide or protein sequences
-
Support for identifying fragments from multiple oligo or protein sequences (e.g., duplexes)
-
Support for specifying modified oligonucleotides
-
Support for “fuzzy” target masses such as PTM’s or protein drug conjugates
-
Dynamic sorting of columns in web-based reports
-
Target mass purity estimation computed from deconvolution peak intensities and chromatogram area percents
-
XML output
Additional Features of ProMass HR:
ProMass HR includes all of the features of the standard version of ProMass, plus the following:
-
Includes the full ReView PPL data processing package with ReSpect™ algorithms
-
Deisotoping for monoisotopic mass determination of isotopically resolved mass spectra for superior mass accuracy (e.g., <3 ppm on Orbitrap data).
-
Mass spectral peak shape modeling for enhanced resolution of isotopically unresolved mass spectra
-
PPL charge deconvolution algorithm for isotopically unresolved mass spectra
-
For more information, access our ProMass HR applications guide.
ProMass for Xcalibur System Requirements
You should follow the general system requirements for installing the version of Xcalibur that you plan to use. The system requirements for running ProMass for Xcalibur are listed below:
-
Thermo Xcalibur v. 2.0.7 or later must be installed
-
The Thermo XDK (included on the Xcalibur CD) should be installed to enable the automation features
-
The required PC has the same requirements of the installed version of Thermo Xcalibur
-
Microsoft Windows XP through Windows 10 (limited by Xcalibur-Windows compatibility)
-
At least 110 MB of free disk space is required to install the program and help files
Spectral deconvolution is a processor-intense operation. Therefore, a fast microprocessor is best if you want fast execution times or if you are processing complex spectra.
Note:ProMass does not include web server software. A web server is not required if you want to view your results on a local machine. However, if you want to share your data with others over the internet or intranet you will need a web server.


- 2026-07-24
- 2026-07-21
- 2026-07-15
- 2026-07-15
- 2026-07-03
- 2026-06-30

- 2026-07-24
- 2026-07-24
- 2026-07-24
- 2026-07-24
- 2026-07-23
- 2026-07-23











