



Q-Chem - 量子化学程序包
Q-Chem软件是一款高精度量子化学计算工具,支持HF/DFT以及各种post-HF计算方法,在理论化学、材料化学、生物化学等相关领域的研究和生产中发挥了很大作用。量子化学方法已被证明对研究分子的化学和物理性质很有价值。所述Q-Chem软件汇集了各种计算方法和工具从头计算软件包,大大提高了速度和计算精度。此外,Q-Chem将容纳比以前更大的分子结构,不会损失精度,从而将量子化学的力量带到以前无法使用该工具的关键研究项目中。这款量子化学软件包,用于预测分子结构,反应性以及振动,电子和NMR光谱。
Q-Chem是由多个研究机构共同开发的从头计算量子化学软件包,开始是由诺贝尔化学获得者John.A.Pople主导开发的。从1999年Q-Chem1.2发布至今,Q-Chem软件已经成为一个功能很强的量子化学计算程序。目前新的版本是Q-Chem 6.4。
Q-Chem的特点:
-
IQmol是Q-Chem的配套可视化软件,可以领取。Q-Chem也支持第三方可视化软件,如Avogadro、WebMO、Jmol等。
-
可进行跨节点并行计算。程序的代码经过完善,拥有更快的计算速度。
-
支持多种方法分析势能面。如冻结带方法、从头算分子动力学、路径积分蒙特卡洛方法及偏Hessian分析。
-
可进行能量组成分析。如可以把能量分解成库伦项、交换项及电荷转移项。
-
可研究电子转移耦合及激发能。支持的方法有限制性DFT、直接耦合及分子片段电荷。
-
拥有不一样的基态泛函。如色散校正泛函XDM、DF-04、XYGJ-OS、WB97X-2等。
-
快捷和更加准确的MP2方法。如SOS-MP2、双基组MP2及局域MP2方法。
-
支持激发态研究(包括开壳层分子)。如SOS-CIS(D)、TDDFT、ADC等。
-
Q-Chem 提供个人技术服务,并会依据客户的要求开发所需的功能。
功能:
基态自洽场方法
1. Hartree-Fock方法
限制性,非限制性,和限制性开壳层形式
用于结构的解析一阶导数
用于谐振频率分析的解析二阶导数
2. 密度泛函理论
局域泛函和梯度校正泛函。交换泛函:Slater,Becke'88,Perdew'91,Gill'96,Gillbert-Gill'99,Handy-Cohen OPTX。关联泛函:VWN5,Lee-Yang-Parr,Perdew-Zunger'81,Perdew'86,Wigner,Perdew'91。EDF1交换-关联泛函。用户定义的交换-关联泛函。
HF--DFT混合泛函:B3LYP,B3PW91,B3LYP5,用户定义的混合泛函。
局域数值格点的数值积分方案:SG-0标准网格,SG-1标准网格,Lebedev和Gauss-Legendre角向积分方案
用于结构的解析一阶导数
用于谐振频率分析的解析二阶道速
3. 线性标度方法
傅立叶变换库仑方法
连续多级方法
线性标度HF交换方法
基于格点的线性标度积分,用于交换—关联泛函求值
线性标度NMR化学位移
4. AOINT包用于双电子积分
结合了高性能积分技术的新进展:COLD PRISM;J-矩阵引擎
SCF改进
in-core和直接SCF的混合
DIIS
初始猜测方案:重叠球平均原子密度,广义Wolfsberg-Helmholtz,从小基组投影,芯哈密顿量的猜测
SCF波函的分析
重叠方法
Fock矩阵的直接小化
计划原子轨道对分子的小基
基于波函的电子关联处理
1. M∅ller-Plesset围绕理论
限制性,非限制性,和限制性开壳层形式
直接和半直接方法计算能量
半直接方法的解析梯度,用于限制性和非限制性形式
在MP3,MP4和MP4SDQ方法的解析梯度计算中处理冻芯轨道
2. 局域MP2方法
根据物理图像截断MP2的能量表达式,从而减少计算量
减少计算量相对分析尺寸的标度,近似为两倍,却不明显丢失精度
应用外推PAO用于局部校正
可使用分子中的双原子和分析中的三原子技术
3. RI-MP2
比MP2和局部MP2快十倍
4. 耦合簇方法
CCSD:能量,以及作为能量有限差分的梯度
EOM--CCSD;=EE,EA,IP,SF,能够处理自由基,键的断裂,以及对称破缺问题
耦合簇能量的非迭代校正:三级校正CCSD(T),三级和四级校正CCSD(2)
广泛应用分析点群对称性,以效率
二次双激发耦合簇
QCISD,QCISD(T)和QCISD(2)用于能量
DIIS用于收敛加速
冻芯近似,用于增加可处理体系的尺寸
5. 轨道的耦合簇方法
轨道的双激发耦合簇(OD):可避免人为的对称破缺问题;平均长参考轨道使能量变小;Brueckner耦合簇;OD,OD(T),和OD(2)的能量及梯度
价轨道的耦合簇方法(VOD):传统CASSCF方法的耦合簇近似;在价活性空间利用截断的OD波函;比CASSCT有更少的磁盘空间需求和更小的体系标度,可处理较大体系;VOD,VOD(T),VQCCD,和VOD(2)的能量及梯度。
激发态方法
1. 计算类型
垂直激发吸收谱
通过激发态能量的有限差分,进行激发态的结构
UCIS和RCIS进行激发态的振动分析
自旋反转DFT
2. CIS方法
从Hartree-Fock基态波函计算激发态:获得定性的单电子激发态;结构与频率与基态Hartree-Fock结果有可比性
的直接算法用于计算闭壳层和开壳层体系的能量、解析梯度和二阶导数
XCIS用于二重和四重态计算
双激发微扰校正CIS(D),可使CIS误差减少两倍或更多,接近于MP2
3. TDDFT
从Kohn-Sham基态波函计算激发态能量
对于低位价激发态,TDDFT比CIS有相当大的计算量
提供激发态中关联效应的内在图像
自由基的低位价激发态,比CIS有相当大的
自旋反转密度泛函理论(SFDFT):把TDDFT推广到低位价激发态之外;可用于键断裂的过程,以及自由基和双自由基体系
4. 基于耦合簇的激发态方法
EOM-CCSD
自旋反转激发态方法:了双、三自由基系统的处理;结合单行列式波函处理键断裂问题;可用于OD和CCSD理论
OOD方法:与CCSD激发态方法有几乎相同的数值性能;比TDDFT精度,到计算量更昂贵
EOM-VOOD方法:于EOM-CCSD,但使用VOOD方案
激发态计算:跃迁偶级矩和结构
5. 分解分析
显示电子跃迁的工具,用于把电子跃迁分类为价跃迁、Rydberg跃迁,混合跃迁,或电荷转换
分析
1. 自动结构和过滤态
使用Jon Baker博士的OPTIMIZE程序包,用约化内坐标迅速收敛,避免初始力常数矩阵
具有约束的结构:可施加于键角,二面(扭转)角,或平面外的弯曲;直角坐标中冻结原子;约束不一定要加在初始结构上。
使用笛卡尔,Z-矩阵或离域内坐标
本征矢跟踪算法,用于过渡态和小化
GDIIS算法用于小化:使到平衡结构的收敛获得很大加速
内反应坐标跟踪;沿着反应路径的连续平衡结构和过渡态
2. 振动光谱
自动调用解析和数值二阶导数
红外和拉曼强度
输出标准的统计热力学信息
同位素替换,用于与实验进行比较
非谐性校正
3. NMR屏蔽张量
4. 自然键轨道分析
使用NBO 4.0
5. Stewart原子
从分析密度重新获得原子
Q-Chem用单位分解方法计算这些值
6. 动量密度
7. Intracules
不一样的双电子函数,提供分子中库仑能和交换能关于位置和动量的详尽信息
8. 分析中的原子
利用AIMPAC进行AIM分析
9. 溶解模型
的Onsager反应场模型
Langevin模型
SS(V)PE:一种新的电解质连续模型
10. 基于Dirac-Fock理论的相对论能量校正
11. 对角热校正
计算Born-Oppenheimer对角修正,研究核与电子运动热距离的分解
基组
1. 高斯基组
2. 赝势基组
3. 用户定义的基组和赝势
4. 基组重叠误差(BSSE)校正
QM/MM
1. 到CHARMM的接口
2. ONIUM
Q-Chem 应用领域:
势能面扫描
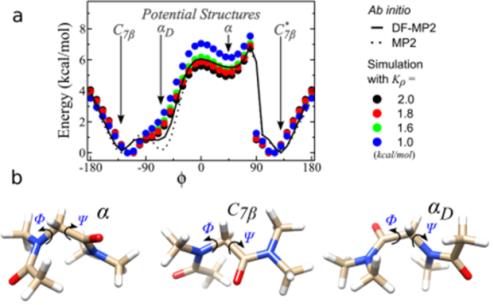
Q-Chem 能够的给出分子在不同构型下的能量,帮助我们找到分子局部的能量很小值以获得稳定的分子结构。还可以根据分子的构象进行势能面扫描。
下图给出了采用Q-Chem计算的di-Peptoid在不同构象下的能量变化。研究者可以通过拟合分子能量和构象的关系构建势能面或分子力场。
分子间相互作用
Q-Chem采用Hartree-Fork,基于泛函的DFT方法和高精度的电子相关方法,多体微扰的MPn与Coupled-cluster理论来计算分子间的相互作用,并可以便捷的进行BSSE矫正。此外,Q-Chem还可以基于ALMO方法将分子之间的相互作用能进行分解。
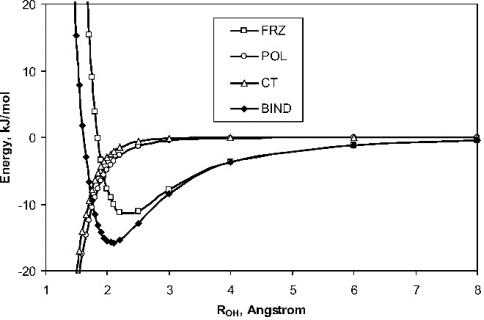
Unravelling the Origin of Intermolecular Interactions Using Absolutely Localized Molecular Orbitals.J.Phys. Chem. A. (2007), 111, 8753
过渡态搜索和反应路径分析
Q-Chem 是业界公认的研究化学反应的有力工具。Q-Chem多种过渡态搜索的方法,并可进一步对反应路径进行分析和。得益于Q-Chem软件的率,研究者可以方便的研究化学反应相关问题。
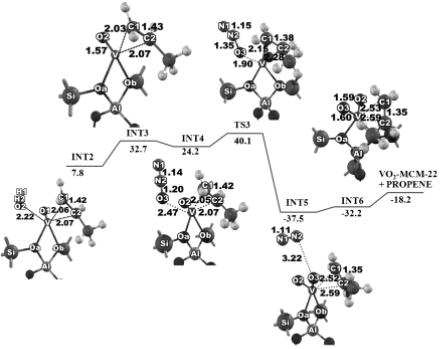
Oxidative Dehydrogenation of Propane over a VO2-Exchanged MCM-22 Zeolite: A DFT Study. ChemPhysChem (2010) , 11, 3432
激发态和光谱计算
除了研究基态的化学性质外, Q-Chem还激发态结构的能量计算和,新版本的Q-Chem还加入了M11等新的泛函并EOM-CCSD方法。Q-Chem可以计算光谱性质,:IR、Raman、紫外-可见、VCD、ECD等。下面的图片就是采用Q-Chem计算的DNA氘化碱基对的红外谱。
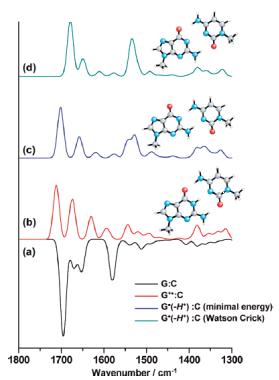
Infrared Characterization of the Guanine Radical Cation: Finger Printing DNA Damage. J. Phys. Chem . B. (2010),114,3660.
线性标度的量化计算
Q-Chem多种线性标度的量子化学反应,:傅立叶变换库伦方法,线性标度HF交换方法,基于格点的线性标度积分等。在下图所示的工作中,研究者采用Q-Chem软件中线性标度的GIAO-HF方法完成了含有1000多个原子体系的NMR化学计算。
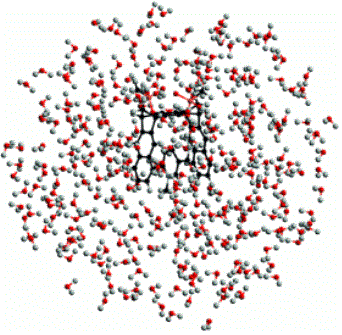
Ab Initio NMR Spectra for Molecular Systems with a Thousand and More Atoms: A Linear-Scaling Method. Angew. Int. (2004), 43, 4485.
QM/MM计算
Q-ChemONIOM计算并自带了与分子动力学模拟软件CHARMM的程序接口。可以方便的进行QM/MM计算。
下图所示的研究工作采用Q-Chem和CHARMM软件考察了钙和镁离子在蛋白磷酸化酶中的作用机理。

Calcium inhibition of Ribonuclease H1 Two-Metal Ion Catalysis. J. Am. Chem. Soc. ASAP.


- 2026-07-29
- 2026-07-28
- 2026-07-26
- 2026-07-24
- 2026-07-23
- 2026-07-21

- 2026-07-30
- 2026-07-29
- 2026-07-29
- 2026-07-29
- 2026-07-29
- 2026-07-29





