



SimMet - 高通量信息学软件
SimMet是一款高通量信息学软件,用于对MS代谢物数据进行定性和定量分析。它使您能够以批处理模式使用MS和MS/MA数据运行代谢物鉴定研究。
SimMet处理LC-MS数据以进行峰检测、峰拾取和保留时间校准。它使用户能够在批处理模式下使用MS和MS/MS数据识别代谢物。除了对已鉴定的代谢物进行定量数据分析外,它还有助于对生物样品中的代谢物进行差异分析。
强的关系代谢物数据库
SimMet数据库包括来自人类、大肠杆菌等各种生物来源的68,459种代谢物。并支持来自NIST MS/MS数据库的9390种代谢物的2,34,284个MS/MS谱图和354个加合物。其他生物信息,如代谢物通用名称、系统名称、质量、组成和与其他数据库的链接,也可供参考。
SimMet通过观察到的MS/MS谱图与代谢物的标准MS/MS谱图进行匹配,提供代谢物的准确鉴定。
支持多种文件格式
SimMet接受标准文件格式的实验质谱数据,例如.txt、.xls、.mzData和.mzXML。它可以读取Thermo Scientific(*raw)和SCIEX(*.wiff)以及Bruker Corporation的(.baf、.yep和.fid)原生数据文件。该程序能够从色谱运行(即多达200,000次扫描)中导入数据。
为您的全部分析提供单一平台
SimMet的综合平台无需管理和使用多种代谢物研究工具。全部代谢物数据分析,从LC-MS数据处理和随后的峰识别到统计分析(如主成分分析(都可在单个工作区中进行。支持加载和得分图以及置信椭圆(相关加载和Hotelling的T2椭圆),以帮助研究人员了解代谢物和样品之间的关系。还提供了代谢物响应曲线以显示不同样品中代谢物丰度的变化。
轻松管理大数据
SimMet旨在有效处理大量数据,这是基于质谱的代谢组学工作流程的典型特征。用户可以在单个项目中加载50万次扫描,以及一次导出50,000次扫描的分析结果。支持全部标准供应商格式,以集成定性和定量工作流程解决方案。
SimMet特点:
LC-MS和MS/MS数据处理
SimMet支持LC-MS数据处理技术,例如峰检测、拾取和识别。数据以批处理模式自动处理,以生成指定保留时间范围内检测到的代谢物表。该工具自动将全部m/z值、丰度、同位素水平、电荷状态和MS/MS数据制成表格。SimMet自动将检测到的化合物与相应的MS/MS谱图对齐,以便基于准确质量搜索以及前体、产物离子和谱图匹配进行准确的代谢物鉴定。
SimMet通过饼图、条形图提供代谢物分布的直观图解释,并使用频率表对样品间的代谢物分布进行比较分析。
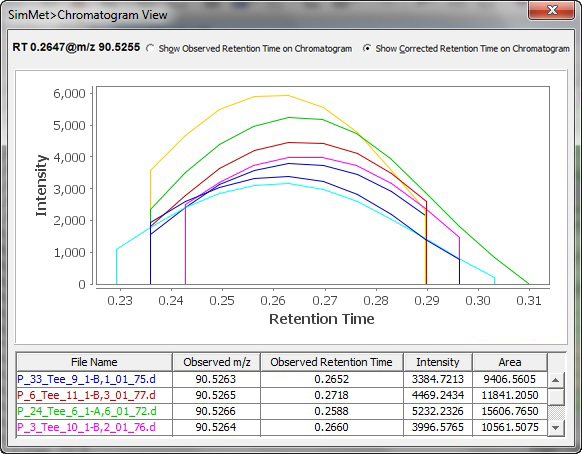
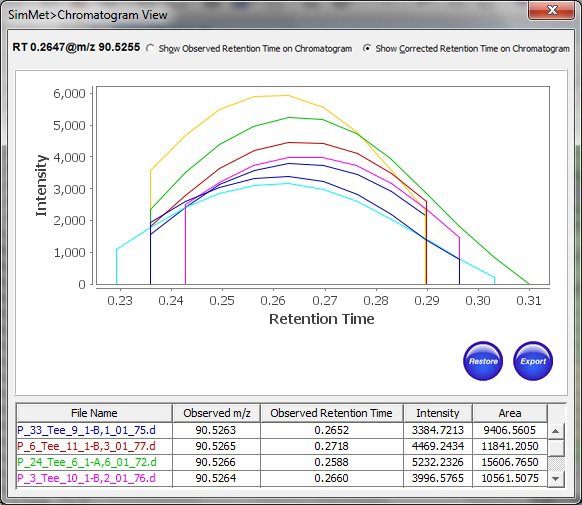
此外,SimMet根据保留时间、电荷状态、概率、分数和模式分数对生物样品中的代谢物进程比对,以方便对分散在不同扫描或配置文件中的数据进行调查。该程序显示MS和MS/MS水平的TIC(总离子色谱图)、XIC(提取离子色谱图)和BPC(基峰色谱图)。色谱图可以放大、以便调查和突出紧要的峰。可以导出绘图以在实验室中共享结果。
高通量MS和MS/MS数据分析
SimMet通过将观察到的代谢物前体离子和产物离子与SimMet数据库中可用的已知代谢物光谱/结构/片段进行匹配,从而实现代谢物鉴定。SimMet提供直观的用户界面,除了在同一工作空间中显示已识别的2D结构、碎片离子、注释的MS或MS/MS光谱以及已识别代谢物的其他信息外,还可以轻松执行此类分析。
SimMet在一次搜索运行中分析10,000次扫描的MS/MS数据。可以分析具有百万分之1-50(ppm)和0.1到2000 millidaltons(mDa)之间误差容限的高分辨率准确质量数据。使用化合物鉴定和光谱模式匹配算法对结果进行排序,该算法将理论代谢物与实验数据的接近程度映射到一起。
生物样品的差异代谢组学
SimMet中代谢物的主成分分析(PCA)提供了生物样品和代谢物之间关系的可视化表示。研究人员可以轻松分析各种生物样本之间的异同。主成分分析(PCA)生成两个二维图,即分数图和负荷图。为了查明可能的生物标志物,二维图支持置信椭圆,例如相关载荷和Hotelling的T2椭圆。

定量代谢组学
SimMet使用由稀释系列和内标构建的校准曲线对样品中存在的代谢物进行定量数据分析。SimMet使用回归拟合线图方法预测样品中分析物的量。
已鉴定代谢物的质谱注释
SimMet可以使用从MS和MS/MS数据中鉴定的代谢物对质谱进行注释。镜像图也可用于显示已识别代谢物的观察与标准光谱。这有助于通过突出显示与产品数据库中的理论代谢物结构相匹配的实验m/z值来解释质谱。每个匹配的峰都可以用代谢物片段进行注释。带注释的光谱可以作为图像(JPEG或.PNG)导出到MS PowerPoint,以促进研究小组之间的信息共享。注释的质谱可以重新调整以适合页面。或者,用户可以通过指定m/z范围或通过选择图上的范围并导出结果来放大或缩小图。
系统要求:
| 对于Windows | 需要 | 推荐 |
| CPU | Core i3 | 更高 |
| 内存 | 8GB可用RAM | 更高 |
| 硬盘空间 | 100GB可用硬盘 | 更高 |
| 屏幕分辨率 | 1024×786 | >1024×768 |
Windows支持的平台:Vista/Windows 7/Windows 8/Windows 10
【英文介绍】
SimMet is a comprehensive software suite for mass spectrometry metabolite data analysis that facilitates LC-MS data processing, metabolite identification, quantification and statistical analysis.
SimMet processes LC-MS data for peak detection, peak picking and retention time alignment. It enables users to identify metabolites using MS and MS/MS data in batch mode. It enables quantitative data analysis of identified metabolites as well as the differential analysis of metabolites across biological samples.
Robust Relational Metabolite Database
The SimMet database includes 68,459 metabolite species from various biological sources such as Humans, E.coli. and supports 2,34,284 MS/MS spectra with 354 adducts for 9,390 metabolites from NIST MS/MS databases. Additional biological information such as metabolite common name, systematic name, mass, composition and links to the other databases are also made available for easy reference.
SimMet offers accurate identification of metabolites by matching observed MS/MS spectra against standard MS/MS spectra of metabolites.
Support for Multiple File Formats
SimMet accepts experimental mass spectrometry data in standard file formats such as .txt, .xls, .mzData and .mzXML. It can read Thermo Scientific (*.raw) and SCIEX (*.wiff) and Bruker Corporation's (.baf, .yep and .fid) native data files. The program is capable of importing data from complete chromatographic runs i.e up to 200,000 scans.
Single Platform for All Your Analysis
SimMet's comprehensive platform eliminates the need for managing and using multiple tools for metabolite research. All the metabolite data analysis, from LC-MS data processing and subsequent peak identification to statistical analyses such as principal component analysis is available in a single workspace. Loading and score plot coupled with confidence ellipses (correlation loadings and Hotelling's T2 ellipses) are supported to help researchers in understanding the relationship between metabolites and samples. Metabolite response curves are also provided to display the change in abundance of metabolites in different samples.
Manage Large Data Easily
SimMet is engineered to handle massive volumes of data effectively, which are typical of mass spectrometry based metabolomics work flows. Users can load half a million scans in a single project as well as export analysis results of 50,000 scans at a time. All standard vendor formats are supported to seamlessly integrate qualitative and quantitative work flow solutions.


- 2026-07-29
- 2026-07-28
- 2026-07-26
- 2026-07-24
- 2026-07-23
- 2026-07-21

- 2026-07-30
- 2026-07-29
- 2026-07-29
- 2026-07-29
- 2026-07-29
- 2026-07-29












